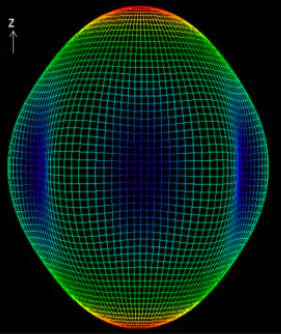
An expression for anisotropic interfacial energy of hexagonal close-packed metals has been formulated which is able to reproduce published data obtained using the modified embedded atom method, covering the variation in interface energy as a function of orientation for a number of metals. It turns out that the coefficients associated with the expression can be determined fully by measured or calculated interfacial energies of just three independent crystal planes. Three-dimensional phase-field model simulations using this representation of interfacial energy have been found to yield convincing crystal morphologies. The apparent rate of crystal growth as a function of orientation in the phase-field simulation agrees with predictions made by surface energy theory.
Acta Materialia, Vol. 57, 2009, 3382-3390.
Download zipped archive of figures
| CML Home | Materials Algorithms |